Our group is engaged in research in the fields of theoretical condensed matter physics and materials science. Our research covers a broad range of materials systems, from bulk materials (metals, semiconductors, and insulators) to those of finite size, such as organic and inorganic nanostructures, to complex multifunctional oxides; phenomena of interest include optical properties, superconductivity, conductance of nanostructures at finite bias, pressure and temperature effects, and dynamics. Particular emphasis is placed on the study of the role of many-particle effects in determining experimentally observed properties.
Our primary goal is to understand and predict materials properties at the most fundamental level using atomistic first principles (or “ab initio”) quantum-mechanical calculations. A variety of different computational approaches are used that require only the atomic number and positions as input. These first principles methods have, in the past, resulted in excellent quantitative agreement with experiment and have predicted with good accuracy materials properties that were later verified experimentally.
We present below some Highlights on recent research projects. For more details, please see our list of publications.
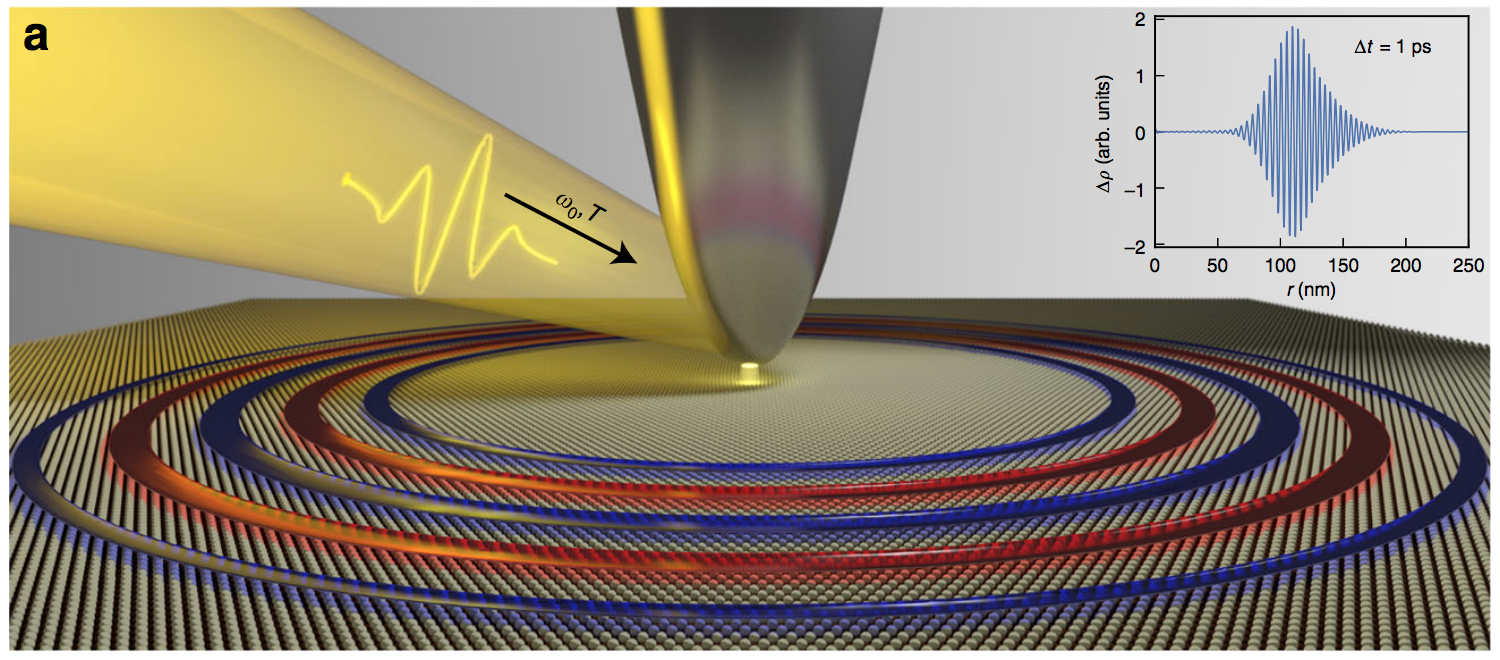
Slow plasmons in atomically thin quasi-2D materials
Plasmons depend strongly on dimensionality: while plasmons in three-dimensional systems start with finite energy at wavevector q = 0, plasmons in traditional two-dimensional (2D) electron gas disperse as square root of q. However, besides graphene, plasmons in real, atomically thin quasi-2D materials were heretofore not well understood. Here we show that the plasmons in real quasi-2D metals are qualitatively different, being virtually dispersionless for wavevectors of typical experimental interest. This stems from a broken continuous translational symmetry which leads to interband screening; so, dispersionless plasmons are a universal intrinsic phenomenon in quasi-2D metals. Moreover, our ab initio calculations reveal that plasmons of monolayer metallic transition metal dichalcogenides are tunable, long lived, able to sustain field intensity enhancement exceeding 10^7, and localizable in real space (within ~20 nm) with little spreading over practical measurement time.
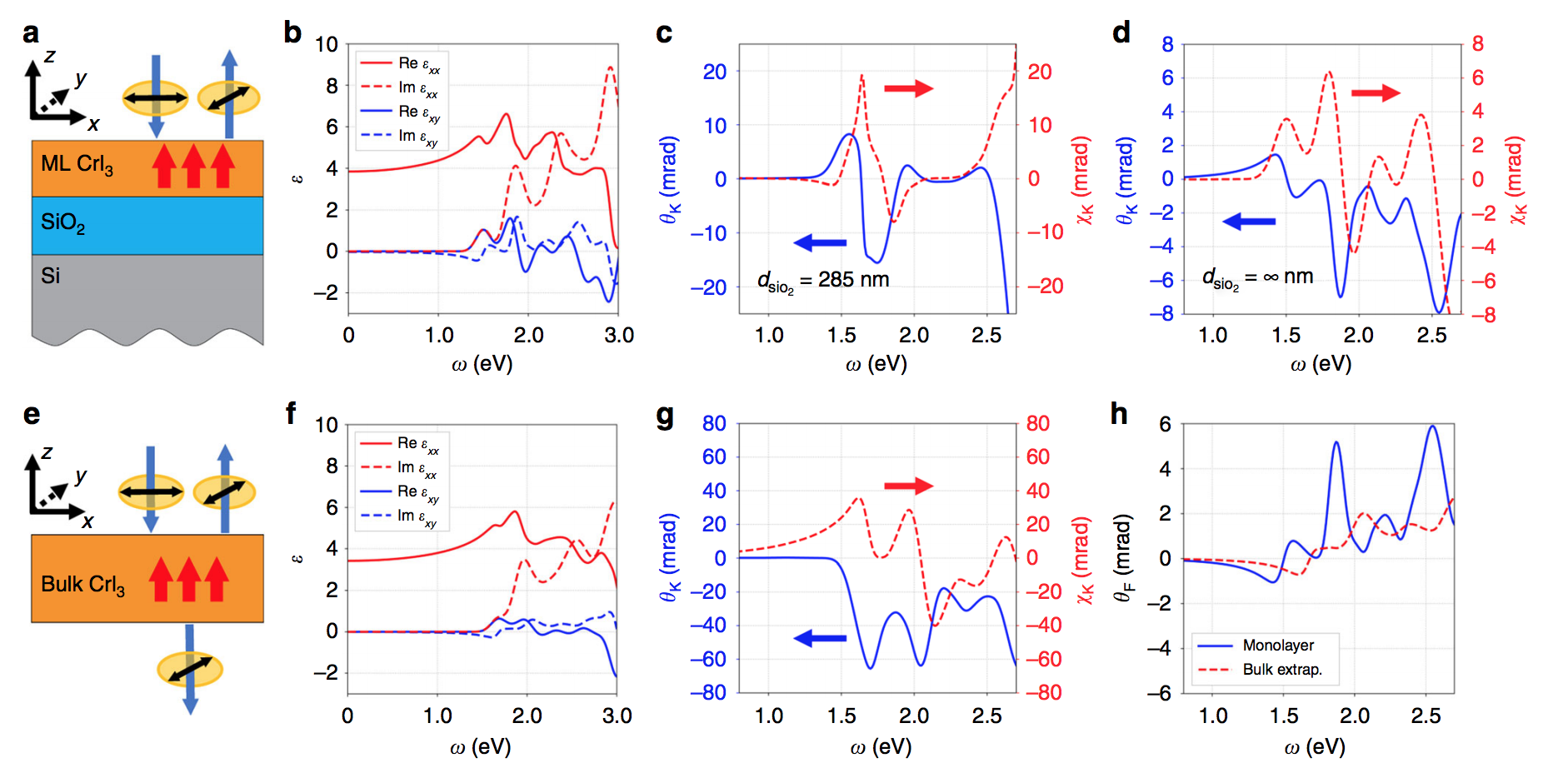
Giant excitonic and magnetooptical responses in two-dimensional ferromagnetic insulators
The recent discovery of magnetism in atomically thin layers of van der Waals crystals has created great opportunities for exploring light–matter interactions and magneto-optical phenomena in the two-dimensional limit. Optical and magneto-optical experiments have provided insights into these topics, revealing strong magnetic circular dichroism and giant Kerr signals in atomically thin ferromagnetic insulators. However, the nature of the giant magneto-optical responses and their microscopic mechanism remain unclear. Here, by performing first-principles GW and Bethe-Salpeter equation calculations, we show that excitonic effects dominate the optical and magneto-optical responses in the prototypical twodimensional ferromagnetic insulator, CrI3. We simulate the Kerr and Faraday effects in realistic experimental setups, and based on which we predict the sensitive frequency- and substrate-dependence of magneto-optical responses.
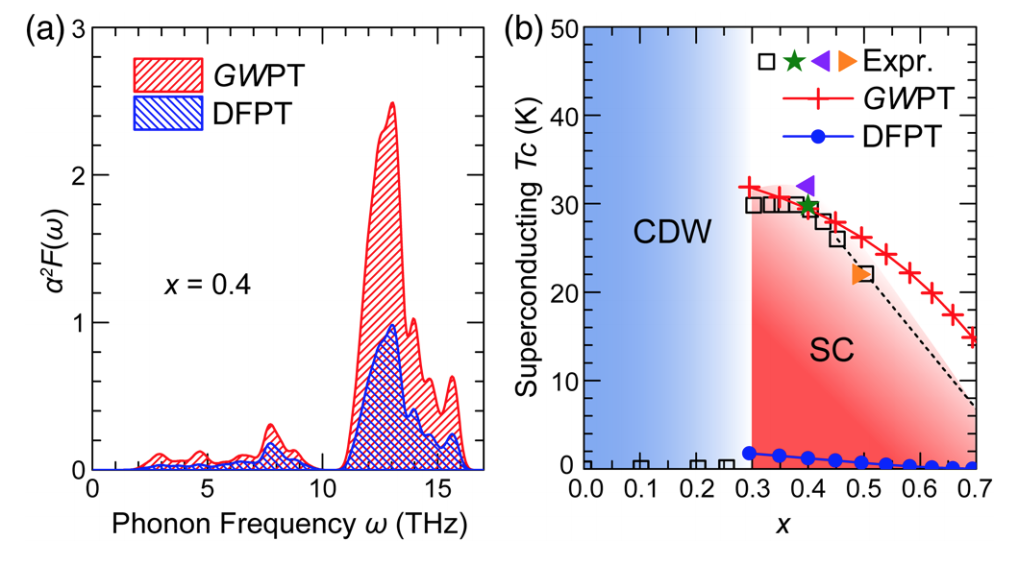
Electron-Phonon Coupling from Ab Initio Linear-Response Theory within the GW Method: GW perturbation theory (GWPT)
We present a new first-principles linear-response theory of changes due to perturbations in the quasiparticle self-energy operator within the GW method. This approach, named GW perturbation theory (GWPT), is applied to calculate the electron-phonon (e-ph) interactions with the full inclusion of the GW nonlocal, energy-dependent self-energy effects, going beyond density-functional perturbation theory. Avoiding limitations of the frozen-phonon technique, GWPT gives access to e-ph matrix elements at the GW level for all phonons and scattering processes, and the computational cost scales linearly with the number of phonon modes (wave vectors and branches) investigated. We demonstrate the capabilities of GWPT by studying the e-ph coupling and superconductivity in Ba0.6K0.4BiO3. We show that many-electron correlations significantly enhance the e-ph interactions for states near the Fermi surface, and explain the observed high superconductivity transition temperature of Ba0.6K0.4BiO3 as well as its doping dependence.
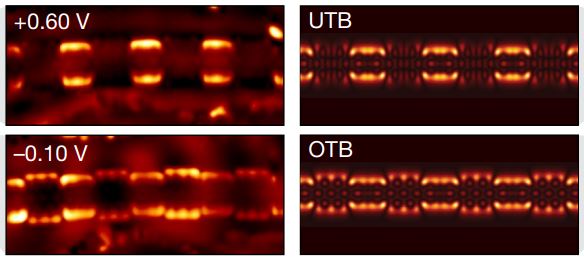
Topological phases in graphene nanoribbons
We predict that semiconducting graphene nanoribbons (GNRs) of different width, edge, and end termination (synthesizable from molecular precursors with atomic precision) belong to different electronic topological classes. The topological phase of GNRs is protected by spatial symmetries and dictated by the terminating unit cell. Our theoretical prediction of junction states and end states arising from the non-trivial topological phases has recently been experimentally observed and confirmed.
- Ting Cao, Fangzhou Zhao, and Steven G. Louie, Phys. Rev. Lett. 119, 076401 (2017).
- Daniel J. Rizzo, Gregory Veber, Ting Cao, Christopher Bronner, Ting Chen, Fangzhou Zhao, Henry Rodriguez, Steven G. Louie, Michael F. Crommie, and Felix R. Fischer, Nature 560, 204 (2018).
- Yea-Lee Lee, Fangzhou Zhao, Ting Cao, Jisoon Ihm, and Steven G. Louie, Nano Lett. 18, 7247 (2018).
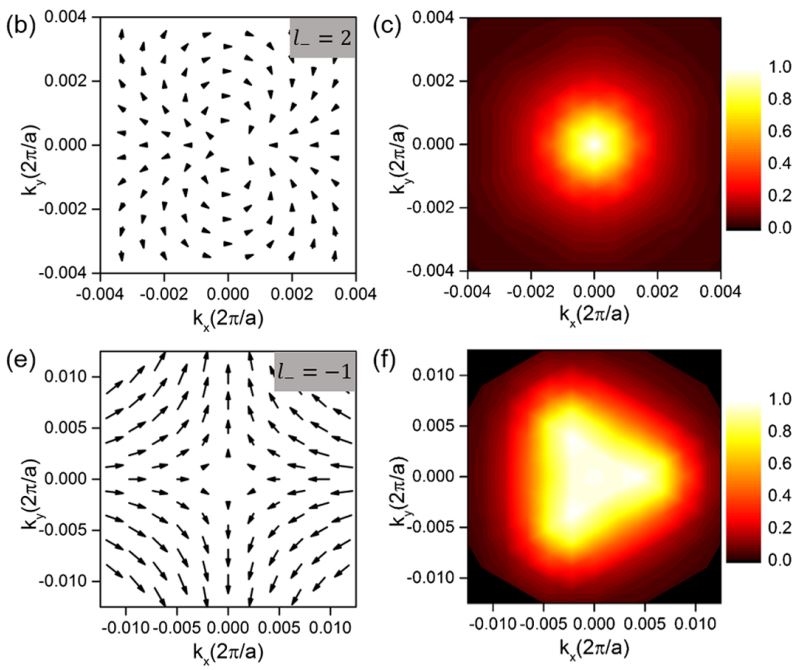
Unifying optical selection rules for excitons in two dimensions: band topology and winding numbers
We show that band topology can dramatically change the photophysics of two-dimensional semiconductors. For systems in which states near the band extrema are of multicomponent character, the spinors describing these components (pseudospins) can pick up nonzero winding numbers around the extremal k point. In these systems, we find that the strength and required light polarization of an excitonic optical transition are dictated by the optical matrix element winding number, a unique and heretofore unrecognized topological characteristic. This winding-number physics leads to novel exciton series and optical selection rules, with each valley hosting multiple bright excitons coupled to light of different circular polarization.
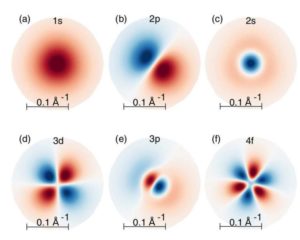
Optical properties of transition metal dichalcogenides and monochalcogenides
Two-dimensional (2D) transition metal dichalcogenides (TMDs) are emerging as a new platform for exploring 2D semiconductor physics. Reduced screening in two dimensions results in markedly enhanced electron–electron interactions, which have been predicted to generate giant bandgap renormalization and excitonic effects. We present first-principles calculations of the optical response of different geometries of molybdenum dichalcogenides, employing the GW-Bethe-Salpeter equation (GW-BSE) approach including self-energy, excitonic, and electron-phonon effects. The resulting optical properties are shown to be dominated by excitonic states with very large binding energy. The optical properties are also strongly related to structures, substrates, and defects.
- D.Y. Qiu, F.H. da Jornada, and S.G. Louie, Phys. Rev. Lett. 111, 216805 (2013).
- Sivan Refaely-Abramson, Diana Y. Qiu, Steven G. Louie, and Jeffrey B. Neaton, Phys. Rev. Lett. 121, 167402 (2018).
- Gabriel Antonius, Diana Y. Qiu, Steven G. Louie, Nano Lett. 18, 1925 (2018).
- K. Liu, L. Zhang, T. Cao, C. Jin, D. Qiu, Q. Zhou, A. Zettl, P. Yang, S. G. Louie, and F. Wang, Nature Communications 5, 4966 (2014).
- D. Y. Qiu, F. H. da Jornada, and S. G. Louie, Phys. Rev. B 93, 235435 (2016).
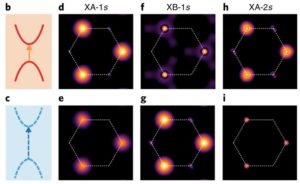
Exchange-driven intravalley mixing of excitons in monolayer transition metal dichalcogenides
Two-dimensional ferromagnetism
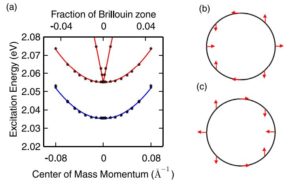
Exciton dispersion in monolayer transition metal dichalcogenides and singlet fission in solid pentacene

Renormalization of quasiparticle and exciton properties induced by substrate screening
We developed ab initio method to include the RPA dielectric effect from substrates to two-dimensional materials to study the renormalization of GW quasiparticle band structures and exciton excitations. We have studied various materials including transition metal dichalcogenides and few-layer black phosphorus, considering the substrate effects from graphene and wide-gap insulators. Our calculations show nice agreement with experiments.
- Miguel M. Ugeda, Aaron J. Bradley, Su-Fei Shi, Felipe H. da Jornada, Yi Zhang, Diana Y Qiu, Wei Ruan, Sung-Kwan Mo, Zahid Hussain, Zhi-Xun Shen, Feng Wang, Steven G. Louie, Michael F. Crommie, Nature materials 13, 1091 (2014).
- Diana Y. Qiu, Felipe H. da Jornada, and Steven G. Louie, Nano Lett. 17, 4706 (2017)
- Likai Li, Jonghwan Kim, Chenhao Jin, Guo Jun Ye, Diana Y. Qiu, Felipe H. da Jornada, Zhiwen Shi, Long Chen, Zuocheng Zhang, Fangyuan Yang, Kenji Watanabe, Takashi Taniguchi, Wencai Ren, Steven G. Louie, Xian Hui Chen, Yuanbo Zhang, and Feng Wang, Nature Nanotech. 12, 21 (2017).
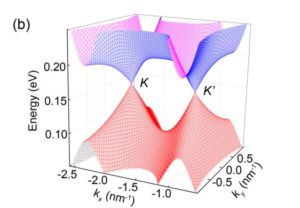
Electronic properties of bottom-up synthesized graphene nanoribbons
Bottom-up synthesis technique has become an important experimental technique that provides large degrees of freedom to synthesize various types of graphene nanoribbons. One important application is bandgap engineering that is used to create semiconductor heterostructure devices that perform processes such as resonant tunneling and solar energy conversion. In collaboration with experiments, we have studied electronic properties of a wide range of graphene nanoribbons that exhibit many intriguing physics.
- Yen-Chia Chen, Ting Cao, Chen Chen, Zahra Pedramrazi, Danny Haberer, Dimas G. de Oteyza, Felix R. Fischer, Steven G. Louie, and Michael F. Crommie, Nature Nanotech. 10, 156 (2015).
- Giang D. Nguyen, Hsin-Zon Tsai, Arash A. Omrani, Tomas Marangoni, Meng Wu, Daniel J. Rizzo, Griffin F. Rodgers, Ryan R. Cloke, Rebecca A. Durr, Yuki Sakai, Franklin Liou, Andrew S. Aikawa, James R. Chelikowsky, Steven G. Louie, Felix R. Fischer, and Michael F. Crommie, Nature Nanotech. 12, 1077 (2017).
- Zahra Pedramrazi, Chen Chen, Fangzhou Zhao, Ting Cao, Giang D. Nguyen, Arash Omrani, Hsin-Zon Tsai, Ryan R. Cloke, Tomas Marangoni, Daniel J. Rizzo, Trinity Joshi, Christopher Bronner, Wonwoo Choi, Felix R. Fischer, Steven G. Louie, and Michael F. Crommie, Nano Lett. 18, 3550 (2018).
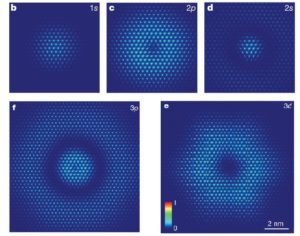
Design novel electronic states in few-layer black phosphorus and graphene
Few-layer black phosphorus has recently emerged as a promising 2D semiconductor, notable for its widely tunable bandgap, highly anisotropic properties, and theoretically predicted large exciton binding energies. We have designed the different forms of black phosphorus to give rise to novel electronic properties such as anisotropic Dirac fermions and gate-switchable transport and optical anisotropy. We have designed several patterned graphene with even disordered potential that gives rise to supercollimation properties.
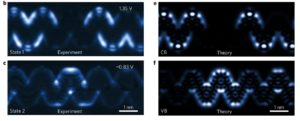
Probing excitonic dark states in single-layer tungsten disulphide
In this work by applying GW-BSE theory, in combination with experimental two-photon excitation spectroscopy, we study a series of excitonic dark states in single-layer WS2. We prove that the excitons are of Wannier type, meaning that each exciton wavefunction extends over multiple unit cells, but with extraordinarily large binding energy (~0.7 electronvolts). These strongly bound exciton states are observed to be stable even at room temperature. We theoretically reveal an exciton series that deviates substantially from hydrogen models, with a novel energy dependence on the orbital angular momentum.
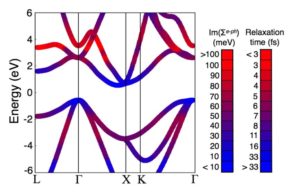
Ab Initio electron-phonon coupling in understanding hot carriers and temperature-induced topological phase transition
Hot carrier thermalization is a major source of efficiency loss in solar cells. We develop and apply an ab initio approach based on density functional theory and many-body perturbation theory to investigate hot carriers in semiconductors. Our calculations include electron-electron and electron-phonon interactions, and require no experimental input other than the structure of the material. We demonstrate that a hot carrier distribution characteristic of Si under solar illumination thermalizes within 350 fs, in excellent agreement with pump-probe experiments. Our work sheds light on the subpicosecond time scale after sunlight absorption in Si, and constitutes a first step towards ab initio quantification of hot carrier dynamics in materials.
- M. Bernardi, D. Vigil-Fowler, J. Lischner, J.B. Neaton, and S.G. Louie, Phys. Rev. Lett.112, 257402 (2014).
- M. Bernardi, J. Mustafa, J. B. Neaton, Steven G. Louie, Nature commun. 6, 7044 (2015).
- M. Bernardi, Derek Vigil-Fowler, Chin Shen Ong, Jeffrey B. Neaton, and Steven G. Louie, Proc. Natl. Acad. Sci. 112, 5291 (2015).
- Gabriel Antonius, and Steven G. Louie, Phys. Rev. Lett. 117, 246401 (2016).

New release of BerkeleyGW 2.0 and recent development in methods and algorithms
BerkeleyGW is a massively parallel computational package for electron excited state properties that are based on many-body perturbation theory employing the ab initio GW and GW plus Bethe-Salpeter equation methodology. We recently released BerkeleyGW 2.0, please visit BerkleyGW website.